









Наприкінці травня стало відомо, що маленькому одеситу Дмитрику Свічинському, який хворіє на рідкісне генетичне захворювання спінальна м'язова атрофія (СМА), таки вдалося зібрати гроші на найдорожчі у світі ліки — укол Золгенсма. Малюк уже його отримав і наразі проходить реабілітацію в США. Дмитрик буде жити: буде повзати, сидіти, ходити, рости… І своїм прикладом дарувати надію іншим хворим дітям, а їх в Україні десятки. То що це за хвороба, що за ліки проти неї та чому вони такі дорогі — читайте в новому матеріалі з рубрики ХЗ (ХтоЗна).
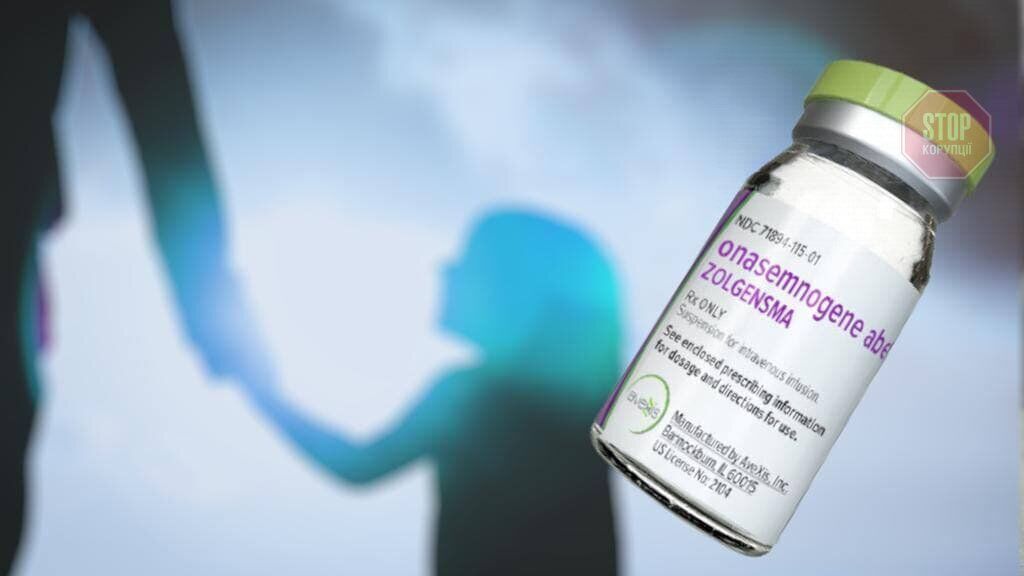
Novartis International AG — це транснаціональна фармацевтична корпорація зі штаб-квартирою у швейцарському Базелі. Упродовж більш як 10 років підрозділи компанії розробляли препарат, який міг би зупинити СМА у дітей, не доросліших за два роки. Якщо на перших етапах одне дослідження відбувалося в одному медичному центрі, то до кінця розробки препарату таких досліджень уже стало до 6 у 20 медичних центрах. У цей час не тільки винаходять самі ліки, а й ліцензують технології, налагоджують виробничі процеси, вишукують майданчик, наймають, навчають і стажують команду спеціалістів, які надалі будуть відповідальними за випуск і доставляння Золгенсма. За більш ніж 10 років клінічних досліджень померло два пацієнти, однак, як повідомляється — не через препарат. Ще десяткам дітей ці дослідження подарували надію: дехто зміг сидіти, а хтось навіть намагається робити перші кроки. Виробництво ліків — процес кропіткий, на певних етапах відбувається вручну, а випуск однієї дози займає близько 30 днів.
Що таке СМА і як працює Золгенсма? Спінальна м’язова атрофія (СМА) – важке моногенне нервово-м’язове захворювання, в основі якого лежать дегенеративні зміни мотонейронів спинного мозку, що прогресують. Захворювання генетичне та поділяється на три типи. Перший — важкий — це найбільш несприятлива форма СМА. Проявляється до шостого місяця життя. Через нестачу моторного розвитку малюки мають труднощі з диханням, смоктанням і ковтанням, такі хворі ніколи не зможуть тримати голівку та сидіти самостійно. Є поняття вродженої форми, яка діагностується у перші дні та тижні життя. Як правило, смерть настає до другого року життя. Другий тип – проміжний – хвороба діагностується з 7-го до 18-го місяця. Дитина може абсолютно нормально розвиватися до етапу самостійного ставання на ноги: вона тримає голівку, сидить, однак надалі не зможе стояти та ходити. Регрес у такій формі настає поступово, при цьому психічний розвиток, зазвичай, відповідає нормі. Смерть настає після двох років. СМА третього типу називають м'якою. У цьому типі захворювання діти встигають опанувати рухові навички, притаманні для 18 місяців. Однак поступово розвивається м’язова слабкість і атрофія, сухожилкові рефлекси регресують. Здатність ходити при цій формі захворювання зберігається до 8-10 років і старше, а самі пацієнти доживають до дорослого віку.
Золгенсма нині — препарат абсолютно революційний, оскільки дозволяє не просто приспати симптоми, а остаточно вилікувати дитину віком до 2 років. При чому за один укол, який забезпечує заміну відсутнього або дефектного гена на його функціональну копію, що призводить до нормалізації вироблення білка виживаності мотонейронів.
Одразу після виходу препарату на ринок почали ширитися домисли, про казкову жадібність виробника, адже ціна за “укол надії” перетинає позначку в два мільйони доларів. Розробники кажуть: жодної жадібності, ціна ретельно вирахувана та відповідає, по-перше, витратам за 10 років досліджень, по-друге, десятирічному курсу терапії за допомогою інших препаратів, і по-третє, складнощам кропіткого процесу виготовлення однієї дози. Крім того, раз на два тижні фармкорпорація розігрує дозу ліків у лотерею для тих дітей, у чиїх країнах Золгенсма ще не схвалили. Загалом до кінця року представники компанії збираються подарувати 100 доз препарату. Цього літа маленький українець із Тернопільщини Арсен Борис виграв таку лотерею.
Між тим дослідження тривають. На сьогодні близько 17 препаратів — у стані розробки на різних етапах клінічних досліджень. Український фонд “Діти зі спінальною м'язовою атрофією” зазначає: СМА є генетичним убивцею №1 у всьому світі дітей віком до 2 років. Крім того, кожен із 40 людей — носій гена, що викликає СМА.
Замість постскриптуму: Дмитрику Свічинському й Арсену Борису надзвичайно пощастило. Однак на сьогодні з десяток українських родин потребують нашої допомоги зі збором коштів, адже державна програма не передбачає лікування таких хворих, а сума в понад 2 мільйони для багатьох є просто непіднімною. Нижче будуть надані всі знайдені мною посилання на групи в соцмережах, створені батьками, яким наша небайдужість може подарувати надію.






